
01 – COMPLEX FORMULATIONS
Material formulations are the result of painstaking, methodical work by countless scientists over decades. Even the humble 2-in-1 shampoo-conditioner took over a decade to develop! (Why? Because shampoo is usually negatively charged, while conditioning agents are usually positively charged; the challenge was figuring out how to prevent these two opposites from attracting each other and consequently inactivating each other). However, as we look forward to replacing the ingredients in formulations with greener, healthier alternatives, we don’t have the luxury of decades to remake our material world. Working with BASF and Dow, I asked: is it possible to use computers to predict how novel molecules self-assemble in formulations?
The scientific challenge is that the reason formulations have interesting properties is usually because small molecules self-assemble into interesting structures. To picture, this, think of the atom in a molecule as an apple-sized object. Now, imagine all of these apple-sized atoms spontaneously organizing themselves and building a skyscraper-sized object. That’s the scale of self-organization we’re talking about. Think of what might go wrong with some bad apples. It turns out that modeling these systems is just as delicate of a matter, and on top of all that models need to span precisely this breadth of scales, from apple to sky scraper.
Our novel contribution was to build low-resolution models from high-resolution models using relative entropy minimization. The resulting low-resolution model is in fact a special kine of field-theoretic model that is uniquely designed to study self-assembly. Consequently, for the first time we calculated self-assembling structures of molecules found in common industrial formulations (surfactants, polymers, polyelectrolytes), completely from theory. These tools are now adopted at BASF, Dow, and the NSF BioPACIFIC Materials Innovation Platform.
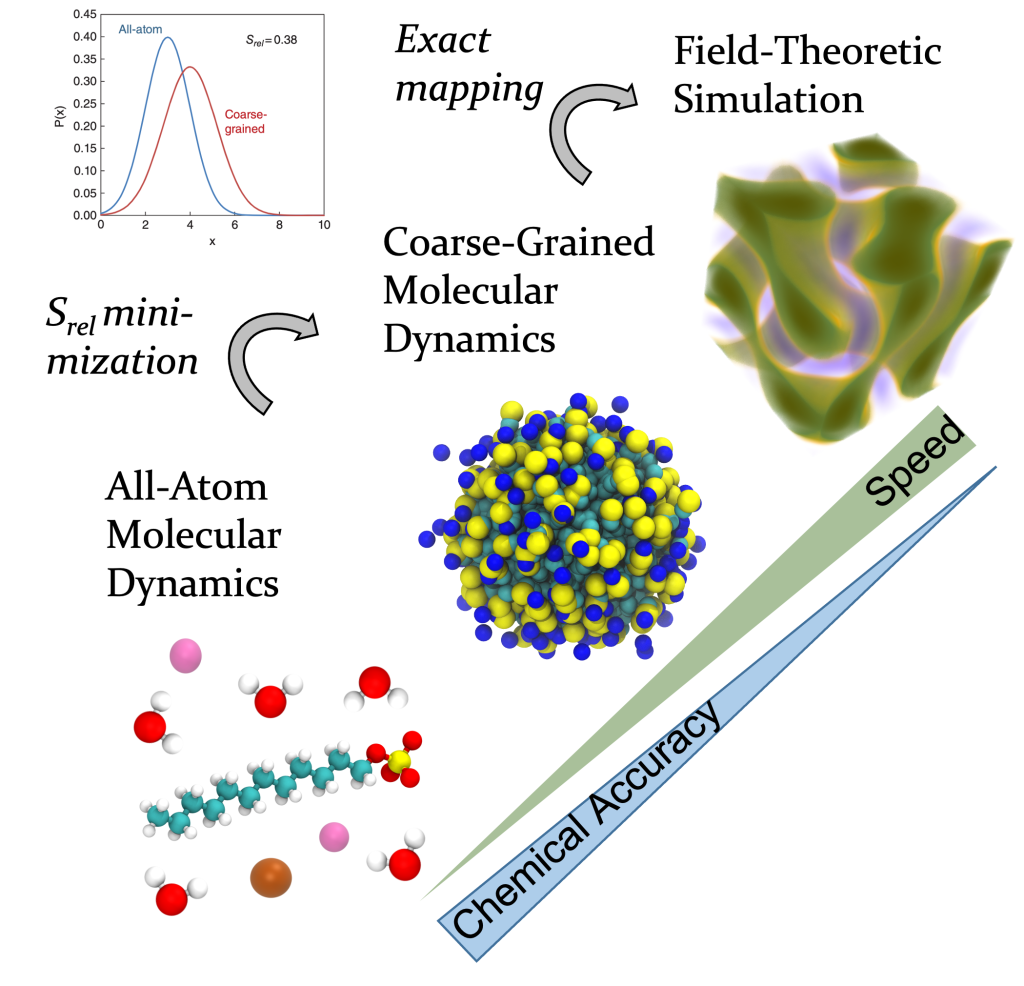
We introduced a novel simulation framework that combines the speed of continuum, field-theoretic simulations with the chemical accuracy of all-atom simulations.
Work:
- K. Shen*, M. Nguyen*, N. Sherck, Sherck, B. Yoo, S. Köhler, J. Speros, K. Delaney, M. S. Shell, G. H. Fredrickson, “Surfactant-polyelectrolyte micelle structure and coacervation transitions studied with a molecularly-informed field theory,” Eur. Phys. J. E 2023, submitted. *Denotes equal contribution.
- K. Shen, M. Nguyen, N. Sherck, B. Yoo, S. Kohler, J. Speros, K. Delaney, M. S. Shell, G. H. Fredrickson, Predicting surfactant phase behavior with a molecularly informed field theory, J. Colloid and Interface Sci. 2023, 638, 84-98
- M. Nguyen, N. Sherck, K. Shen, C. E. R. Edwards, B. Yoo, S. Kohler, J. Speros, K. Delaney, M. S. Shell, G. H. Fredrickson, Predicting Polyelectrolyte Coacervation from a Molecularly Informed Field-Theoretic Model, Macromolecules 2022, 55, 21, 9868–9879.
- N. Sherck, K. Shen, M. Nguyen, B. Yoo, S. Kohler, J. Speros, K. Delaney, M. S. Shell, G. H. Fredrickson, Molecularly Informed Field Theories from Bottom-up Coarse-Graining, ACS Macro Lett. 2021, 10, 5, 576–583.
- K. Shen, N. Sherck, M. Nguyen, B. Yoo, S. Kohler, J. Speros, K. Delaney, M. S. Shell, G. H. Fredrickson, Learning composition-transferable coarse-grained models: Designing external potential ensembles to maximize thermodynamic information, J. Chem. Phys. 2020, 153, 154116 (2020). Editor’s Pick.